Inference of clonal selection in cancer populations using single-cell sequencing data
- Download: You can download the latest version of the software here
- References: TBA
Intra-tumor heterogeneity is one of the major factors influencing cancer progression and treatment outcome. However, evolutionary dynamics of cancer clone populations remain poorly understood. Quantification of clonal selection and inference of fitness landscapes of tumors is a key step to understanding evolutionary mechanisms driving cancer. These problems could be addressed using single cell sequencing, which provides an unprecedented insight into intra-tumor heterogeneity allowing to study and quantify selective advantages of individual clones.
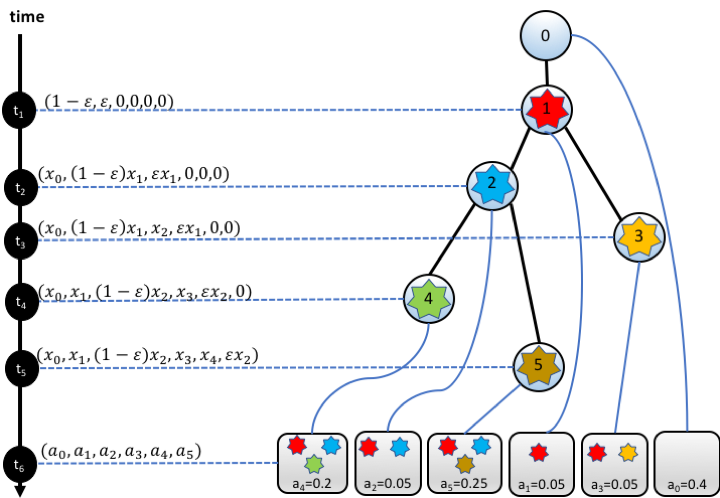
We developed SCIFIL, a computational tool for inference of fitness landscapes of heterogeneous cancer clone populations from single cell sequencing data. SCIFIL allows to estimate maximum likelihood fitnesses of clone variants, measure their selective advantages and order of appearance by fitting an evolutionary model into the tumor phylogeny. We demonstrate the accuracy our approach, and show how it could be applied to experimental tumor data to study clonal selection and infer evolutionary history. SCIFIL can be used to provide new insight into the evolutionary dynamics of cancer.